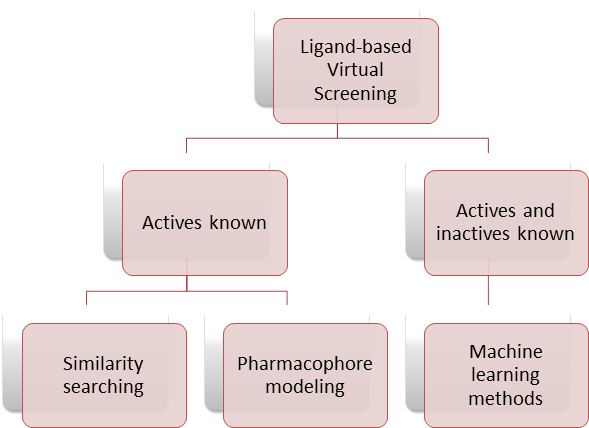
BRUSELAS (Balanced Rapid and Unrestricted Server for Extensive Ligand-Aimed Screening) is a web-based open architecture to perform 3D shape similarity searching and pharmacophore modelling. Both approaches are classified as ligand-based virtual screening (LBVS) methods and are widely used when the 3D structure of the target is unknown. Figure 1. Classification of ligand-based approaches.
This architecture serves to three main objectives:
a) Task automation for virtual screening of a large set of ligands.
b) Ease the use of "in silico" tools to users not familiar with these technique.
c) Reduce time and money expenses related to "in vitro" experimentation.
BRUSELAS can be used to research on a variety of contexts due to its ability to screen large sets of ligands in a few minutes profiting from HPC platforms. It provides a set of parameters to customize tasks including many similarity algorithms, a set of consensus scoring functions and a large collection of descriptors for building on fly libraries. BRUSELAS also maintains a large database of compounds imported from freely accessible databases, including ChEMBL and DrugBank.
Databases available for screening
BRUSELAS maintains a database of 7.473.006 conformers generated from a curated collection of compounds imported from:
KEGG [12]: 23.667 compounds from Compounds and Drugs subsets.
DIA-DB: 186 compounds. DIA-DB is an in-house curated database of antidiabetics.
An average of 8 conformers are available for those compounds containing between 1 and 10 rotatable bonds.
Additionally, BRUSELAS is capable of screening user-provided databases. This feature let users run screening tasks on their own databases.
Screening algorithms
One of the most distinguishing features of BRUSELAS is its ability to use consensus scoring functions to combine different similarity algorithms in one single task. In order to allow the use of many algorithms, BRUSELAS includes the following algorithms:
WEGA [1]. Weighted Gaussian Algorithm (WEGA) significantly improves the accuracy of molecular volumes and reduces the error of shape similarity calculations by 37% using the hard-sphere model as the reference.
LiSiCA [8]. LiSiCA (Ligand Similarity using Clique Algorithm) is a ligand-based virtual screening software that uses a fast maximum clique algorithm to find two- and three-dimensional similarities between pairs of molecules.
Screen3D [9]. A novel, fully flexible shape-based virtual screening algorithm that does not require previous 3D conformation or conformer generation. Due to its solid consistency it can easily be used on desktop computers by non-expert scientists.
OptiPharm. A novel non-deterministic similarity algorithm developed by the University of Almeria.
For pharmacophore modelling, BRUSELAS includes:
SHAFTS [11]. SHAFTS (SHApe-FeaTure Similarity) for 3D molecular similarity calculation and ligand-based virtual screening adopts a hybrid similarity metric combined with molecular shape and colored (labeled) chemistry groups annotated by pharmacophore features for 3D similarity calculation and ranking, which is designed to integrate the strength of pharmacophore matching and volumetric overlay approaches.
Screening workflow
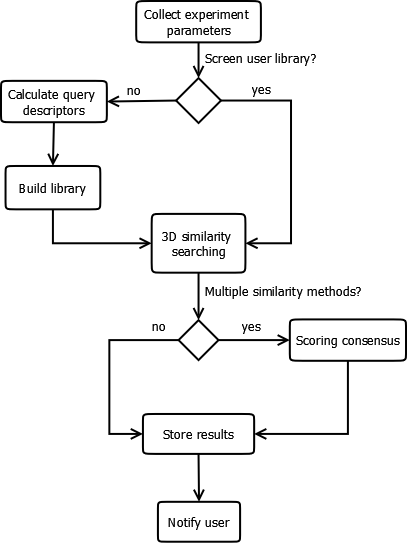
Before we go into details of how tasks can be configured in BRUSELAS, we will briefly explain the way it works. To do so, we will go through the workflow implemented by the architecture to see in detail every step. Figure 2. Screening workflow of BRUSELAS.
First, input parameters are collected from the web interface. Users are be able to choose the most suitable values for their tasks, including the chemical databases to screen and the similarity algorithms to apply. A key point at this stage is whether the users supply their own database or not. If no user database is uploaded, then BRUSELAS generates one on the fly for the current task. To achieve so, the architecture builds a fingerprint based on the molecular descriptors selected by the user. A distance function (e.g. Manhattan, Euclidean) is applied to calculate how similar fingerprints are, and the closest compounds to the query fingerprint become part of the library. In case that a user database be uploaded, it will remain as it is to preserve in its original state.
Once the library is available, the similarity between the query and the ligands in the library is assessed using the selected methods. Every method calculates a similarity score between the query and each ligand in the library. If only one similarity algorithm was chosen, no extra calculation is needed. However, if many of them were picked, a consensus scoring function has to be applied to get the final combined score.
When all the scores are calculated for all the ligands in the library, they are stored in a relational database to make them available for the web tool. Also alignments between query and ligands are calculated and stored.
Finally, users is notified by mail that their results are available on the website.
Step by step screening
In this section we describe how to configure and run a screening task on BRUSELAS step by step.
Configure a task
The configuration of a new task is easy in BRUSELAS because only a few parameters are mandatory. The collection of parameters is splitted into 5 tabs containing options with a similar meaning. Now we will go through those tabs while introducing the set of options contained.
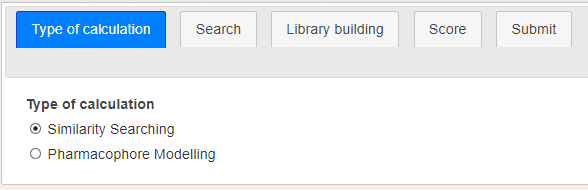
Figure 3. Type of calculation parameters.Figure 4. Second stage parameters.
In the first stage, the user can choose the type of screening to carry out (Figure 3). It can be either shape similarity searching or pharmacophore modelling. The election of one or another will determine the options in the next step. This first stage is mandatory.
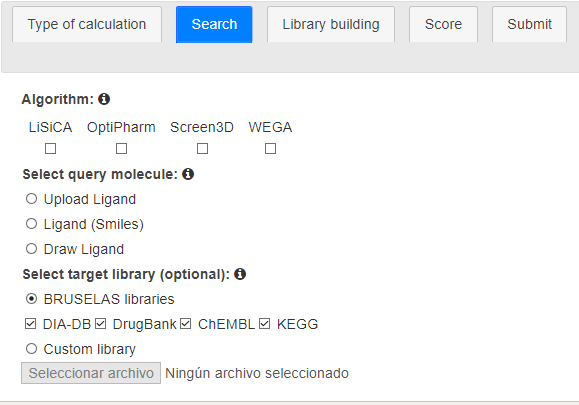
The second stage defines (Figure 4):
The set of similarity algorithms to use. At least one has to be chosen, but many of them may be selected.
The query compound. There are three ways of defining this:
Uploading a file from the user's computer. The accepted formats are the ones admitted by OpenBabel [3] being mol2 the preferred format.
Typing the SMILES representation of the query. If so, a 3D conformer will be generated by BRUSELAS.
Drawing the compound by means the editor.
This is a mandatory input too.
The database(s) to screen. By default, all the databases are selected but users are free to screen the ones they want. They also have the chance of uploading a custom database from a local file.
Figure 5. Building library related parameters.Figure 6. Results related parameters.
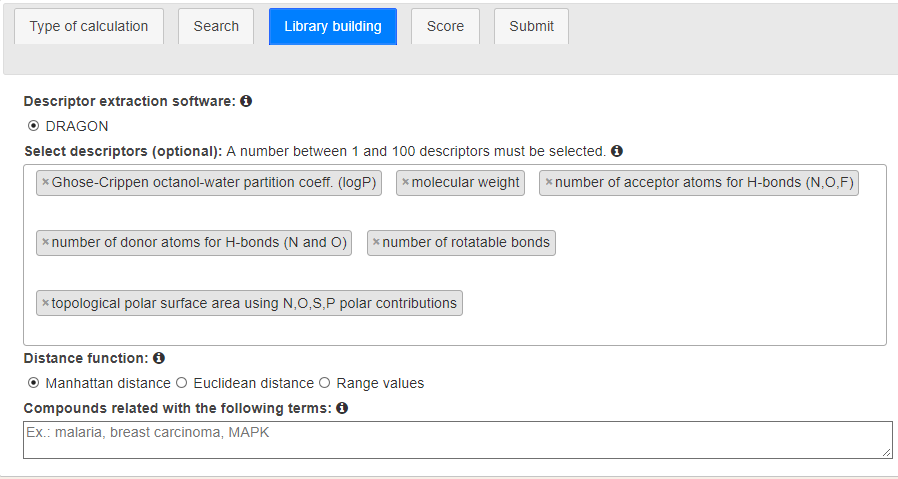
The third stage concerns the creation of a library by BRUSELAS (Figure 6). This step is only useful if the users let BRUSELAS create a suitable library for their task. However, when users decide to screen a custom library, this configuration is omitted.
At this stage, user choose the software used for calculating molecular descriptors. Next, they can select the descriptors that will be considered in the creation of a fingerprint to represent the query and the ligands. A number between 1 and 100 descriptors must be taken. Another important choice is the distance function to measure how similar query and ligands are. Three functions are available (Manhattan, Euclidean and range-based) being Manhattan the default one. Finally, a novel feature on BRUSELAS is the creation of libraries based on key terms. Users can type some terms and only compounds related to such words will be considered in the generation of the library.
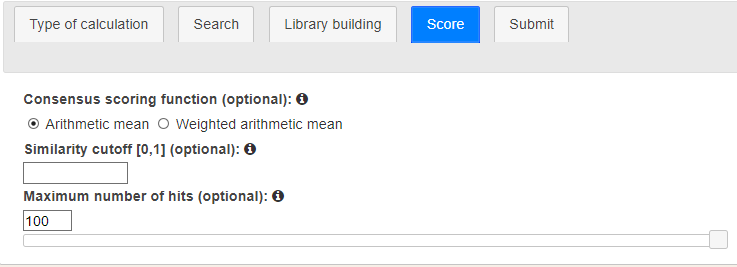
In the fourth stage, users customize the results returned by the architecture (Figure 6). First, a consensus scoring function may be chosen. By default, the average mean is applied on the individual scores but a weighted mean may be calculated too. In that second case, a weight will be given to each score depending on the algorithm that it comes from.
A scoring cutoff is helpful to discard the least promising results. Any compound with a total score under the cutoff will be disregarded. The last option controls the number of results to be returned. A number of results between 10 and 100 must be given (by default 100).
Figure 7. Notification options.
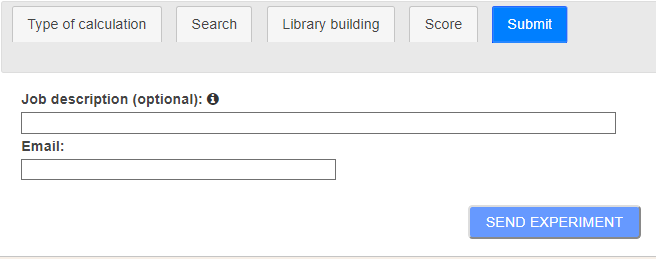
The last stage defines notification related options (Figure 7). A brief text can be attached to the task. That text will be included in the notification mail to identify the task that the user is being informed about. Finally, users must introduce an email address where they want to be notified of the results of the task.
Analyse results
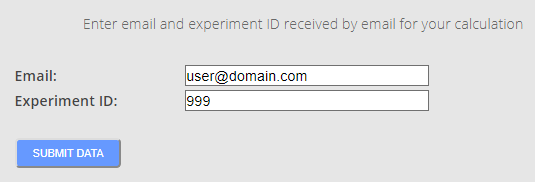
Once the calculation are done the users are notified by email about that. The notification email contains a link to address users to the results screen. To preserve confidentiality of results, the task number and the email provided for its creation are required to access the results (Figure 8).
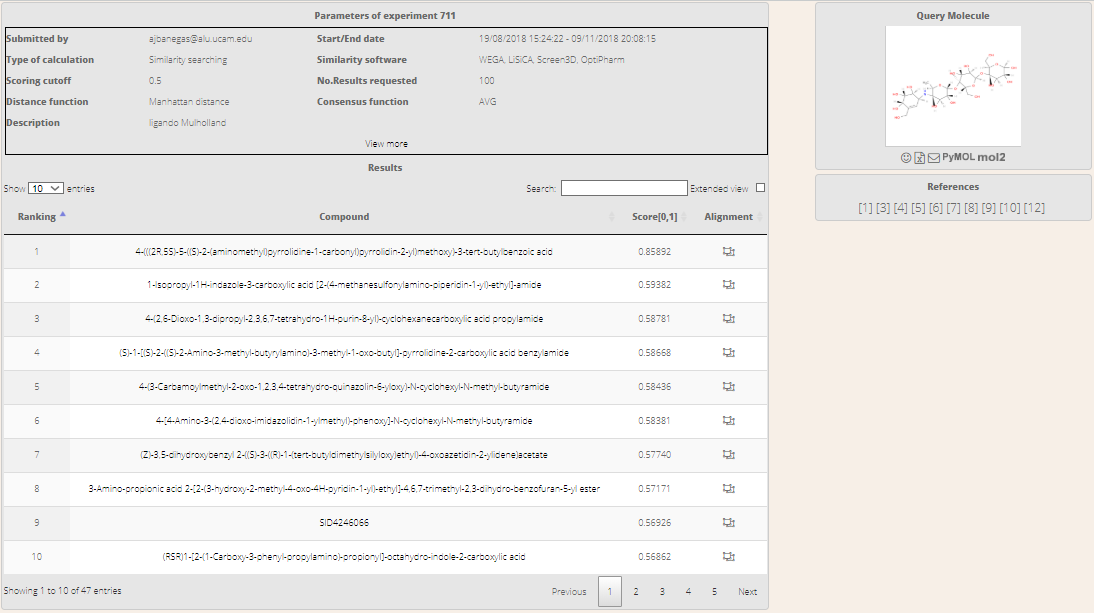
Once autenthicated users will see the list of hits proposed by BRUSELAS along with a summary of the input parameters (Figure 9).
Figure 8. Login screen to access results.Figure 9. List of ranked hits.
The screen is arranged in three blocks:
A header containing a summary of the main input parameters defined in the task creation stage. Only the main parameters are visible initially, but all the other can be verified by clicking on View More.
The main block displays the list of results sorted by descending score. The list includes four columns: i) the rank; ii) the compound's name; iii) the total score; and iv) a link to the alignments screen. This block contains a search input box in the header to filter the list by compound's name. Furthermore, all individual scores can be shown by enabling the Extended View checkbox.
The right side of the screen shows then 2D view of the query and the action buttons just below. If users click on the image, it will be enlarged for a better view. For further details about the actions, please read the following section.
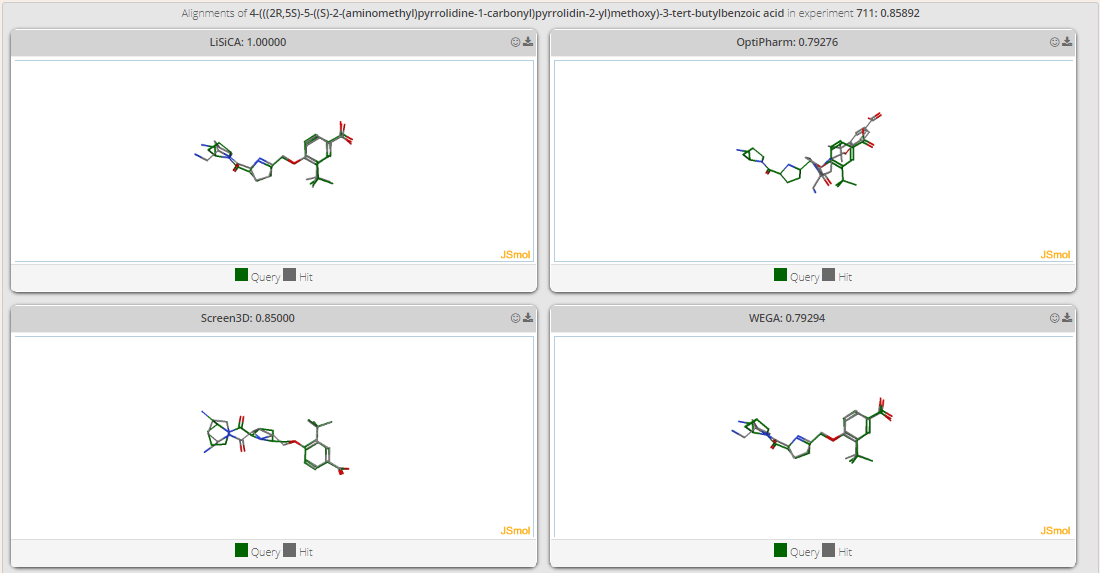
Although scores are indicative of the similarity between the query and ligands, a visual inspection is much more helpful. To achieve so, the alignment screen displays the query overlapped with the ligand according to the calculations of each algorithm (Figure 10). Figure 10. Alignment of query and ligand.
Each alignment is displayed in a box and loaded on Jmol. On top of every box there are two action buttons. The former downloads the SMILES representation of the aligned ligand by the given algorithm. The latter downloads the aligned ligand in a single mol2 file for offline manipulation.
Download results
Figure 11. Actions to perform on the results.
The results of a task can be downloaded in different formats using the action buttons shown in Figure XX. The available actions are:
Download the SMILES representation of the query.
Download the ranks, names and scores in an Excel file.
Share the results page with other users. A link to the page is attached in a mail.
Download the query and all the aligned ligands in a PyMOL session. Only the five best-scored ligands and automatically highlighted. The session is structured in a menu where each entry represents a similarity algorithm. By default, the query is shown along with a second level menu containing the ligands. However, since Screen3D handles flexibility by itself, it is arranged in another way. Each entry in the meny contains the query and the ligand conveniently aligned.
Download the query and all the aligned ligands in mol2 format. A folder with the name of the experiment contains the query and one folder for each algorithm. All this structure is compressed in a .tar.gz file.
Manual search of compounds
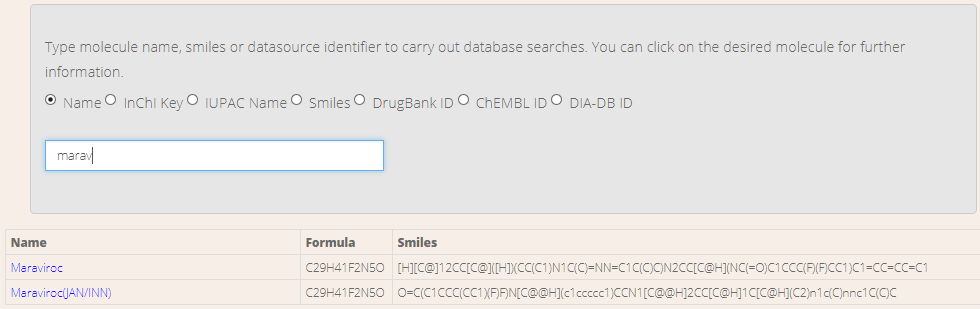
Figure 12. Search screen.
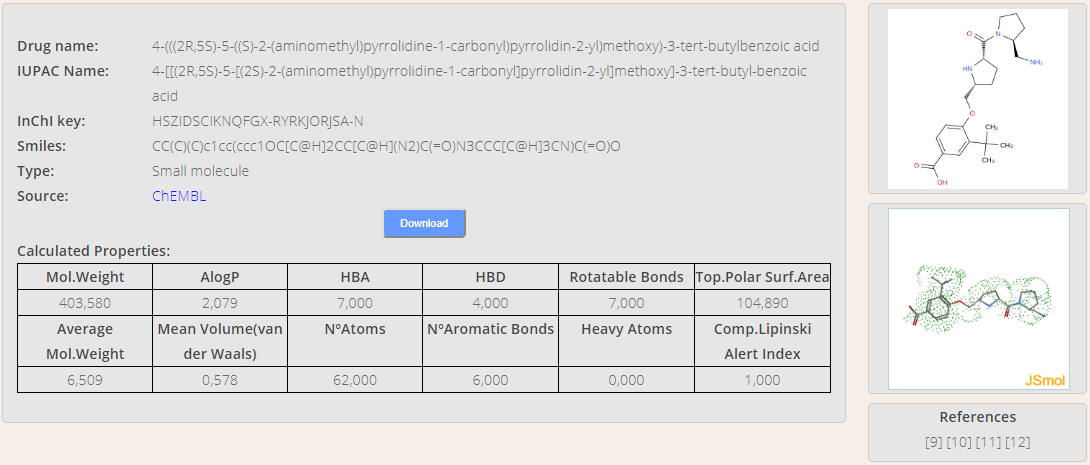
Compounds in the database can be looked for to query extra details. A search engine is available by clicking on Search menu (Figure YY). Users can choose the field that they want to search for. Then, they just need to type part of the pattern to find in the input box and the list of matching compounds is automatically refreshed. Finally, the user will be addressed to the compound's detail card (Figure ZZ) by clicking on the name.
The detail card displays some useful information concerning the compound. Such information includes the chemical name, the IUPAC name, the InChl key, the SMILES chain and the type of molecule. In order to get further details, a link to the source website is provided as well. Moreover, a copy of the compound can be downloaded in mol2 format through the Download button. Figure 13. Compound's detail card.
A preview of compound's shape in 2D and 3D representations is displayed too. The 3D view is loaded in Jmol viewer, therefore the user can play with it.